


Clinical trials testing gene therapies and other gene-based medicines come with operational, regulatory, and biosafety-related considerations that are not required for trials testing most “traditional” drugs. By planning ahead and ensuring investigators are aware of these requirements, gene therapy trial sponsors can dramatically increase the likelihood of conducting a smooth, safe study.

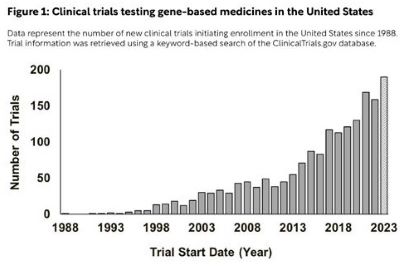
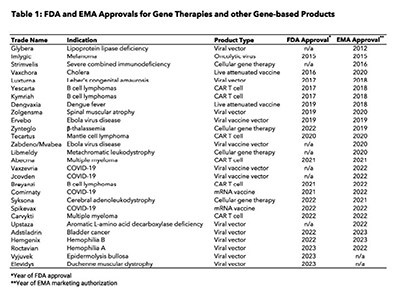
Today there are more than 10,000 known rare diseases, defined by the European Union as conditions that affect less than 1 in 2,000 people and by the United States as those that affect fewer than 200,000 people. The vast majority of rare diseases are caused by or associated with detrimental mutations in protein-coding genes, making them prime candidates for correction with novel gene therapies and other gene-based medicines. Thanks to scientific advances in genetic engineering technologies over the last 20 years, clinical trials testing gene therapies and other gene-based medicines are more common than ever, with nearly 200 opening to enrollment in the United States this year alone (Figure 1). This trend in gene therapy trial starts is not unique to the United States, as similar increases have occurred in Europe and Asia. The European Medicines Agency (EMA) and Food and Drug Administration (FDA) have also each approved more than 10 gene-based medicines for use since 2021 following a long period of relative inactivity (Table 1).
Despite this growth, most trials testing gene therapies and other gene-based medicines fail. While that is also the case for most traditional drugs, gene therapy trials often fail for unique reasons, ranging from unanticipated safety concerns to difficulties in patient recruitment and site-level operations. Understanding the complexities associated with gene therapy trials for rare diseases is critical to planning robust, successful trials that will bring life-changing treatments to patients quickly. This article describes some of the most problematic aspects of conducting a gene therapy clinical trial and outlines strategies that trial planners and sponsors can use to design safe, successful trials.
Unlike many trials where large numbers of healthy participants are desired, trials testing gene therapies for rare diseases are limited by the number of individuals living with the disease. Participant eligibility criteria should therefore be carefully crafted to include populations relevant to the disease in question without unnecessarily excluding patients for reasons unlikely to impact safety or efficacy. Protocols should allow as much flexibility in eligibility criteria and screening processes as possible without compromising participant safety. As gene therapies advance, inclusion of pre-symptomatic patients should also be considered. Depending on the natural course of the disease, administration of the gene therapy before symptoms arise may provide maximal benefit – and in some cases, keep the disease at bay indefinitely. This is especially important for pediatric indications where early intervention is desirable.
Appropriate site selection is vital to planning an effective gene therapy trial. This is largely due to two factors: the limited number of individuals living with the disease within a particular geographic area, and the operational complexities that come with gene therapy trials. For these reasons, sponsors should seek out well-known sites with in-house gene therapy experts, established patient networks, and a history of successfully enrolling patients and completing gene therapy trial activities. Oftentimes a “spoke and hub” approach to site selection is best, where patients are recruited at satellite sites, dosed at a central site, and return to satellite sites for follow-up assessments. If such an approach is used, satellite sites should be conveniently located for patients, and sponsors should plan on reimbursing patients for transportation and lodging costs incurred for travel to the central site. Whatever the case, sponsors should try to set the trial up for success by steering clear of sites that have a history of underperforming on prior trials.
For many rare diseases, enrolling patients at sites in different countries may be necessary to accrue the desired number of participants. And with effective planning, multinational trials can yield valuable data from diverse populations. However, conducting a gene therapy trial in different countries can be very difficult to manage, as clinical trial regulations and healthcare systems can vary widely from one country to another. This, when coupled with inherent cultural and socioeconomic differences between countries, can make it extremely challenging to manage a gene therapy trial in multiple countries.
Choosing and designing a gene therapy vector is one of the most important decisions faced by sponsors and investigators planning new trials for rare diseases. An ideal gene therapy vector should efficiently enter desired cells, drive sustained expression of the corrective transgene, and do so without compromising participant safety. Today, most rare disease gene therapies are based on adeno-associated virus (AAV), which is nonpathogenic and can drive long-term gene expression from non-integrated AAV genomes harbored outside of the nuclei of transduced cells. Engineered capsids and transgene promoters have also emerged as powerful tools that can be used to limit vector transduction and transgene expression to cells and/or tissues of interest.
Compared to more traditional products, virally vectored gene therapies come with unique biosafety concerns related to product shedding. Most viral vectors, like those based on AAV, are unable to replicate within participants after dosing. For these types of vectors, shedding is a small risk. For others, however, this is not the case, and the likelihood of vector shedding and transmission to others is substantially increased. This is especially true for products capable of replicating within participants after dosing, which are most commonly used as oncolytics for the targeted treatment of various cancers.
To mitigate risks associated with product shedding and transmission to others, sponsors should develop clear post-dosing instructions for participants that outline the risks and strategies to mitigate those risks. These instructions should be based on empirical evidence wherever possible, such as preclinical observations or known routes of shedding for similar products in humans. For example, if a product is known to be shed through saliva, participants should be instructed to not share food or drinks with others. Shedding and transmission risks should be noted in informed consent forms (ICFs) as appropriate, and depending on the degree of risk, sponsors should consider creating separate ICFs or information sheets specifically for close contacts of participants, like parents, spouses, and caregivers.
Finally, patients dosed with virally vectored gene therapies will usually mount an immune response against elements of the vector itself that may compromise efficacy of subsequent doses with the same vector type. Study assessments and screening procedures should therefore include evaluation of antibodies against the vector and transgene encoded by the vector at baseline and throughout the study. As gene therapies and similar products become more commonplace, it will be important for trial sponsors to develop novel vectors and/or immunosuppression strategies to get around pre-existing immunity. Directed evolution, library screening, and rationally-designed mutagenesis approaches have all been used in recent years to generate vectors that are antigenically distinct from others and can target specific cell types.
Generally speaking, randomized placebo-controlled studies are the gold standard in clinical trial design, and should be used where possible. However, gene therapy trials for rare diseases are limited by the small number of patients living with the disease who may qualify for the trial. For this reason, placebo controls and other design elements common in larger trials may be impractical or impossible. In the absence of such a placebo-controlled design, appropriate alternatives should be considered. These include using intra-subject controls where appropriate, using historical control groups aligned with the natural history of the disease, and the use of multiple dose level cohorts. Irrespective of the specific design chosen, gene therapy trials for rare diseases should be designed in a way that has the potential to generate safety and efficacy data in support of a marketing application. Beginning with the first-in-human trial, sponsors should also attempt to collect as much participant data as is feasible, including adverse events, efficacy outcomes, and biomarker data that may be used to inform the design of additional trials.
Wherever possible, study endpoints should be clinically meaningful for participants, including those that assess quality of life or other participant-derived subjective measures. In many cases, surrogate endpoints unlikely to occur spontaneously in the natural course of the disease may also be suitable. Examples of appropriate surrogate endpoints include reduced frequency of vaso-occlusive crises for those with sickle cell disease, increased skeletal muscle dystrophin levels for Duchenne muscular dystrophy, and reduced organ volumes for participants with Gaucher disease.
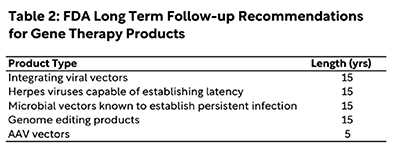
Lastly, most gene therapies are designed to enter cells and drive long-term expression of a corrective transgene, as noted above. While extremely powerful, the sustained activity of gene therapies after dosing increases the risk of delayed adverse effects. This is especially true for products that harbor or drive corrective changes integrated within the cellular genome, as integration or gene editing at the wrong place within the genome – like in an oncogene or tumor suppressor gene – can drive the formation of new cancers. For these reasons, sponsors should design robust long term follow-up plans tailored to the disease and vector being used. Currently, the FDA recommends 15 years of follow-up after dosing with several classes of gene therapy products based on the likelihood of delayed adverse effects arising; similar guidance recommends at least five years of follow-up after dosing with AAV vectors. While these recommendations may sound cumbersome, they are critical to ensuring participant safety and instilling public confidence in the field as gene therapies advance. The FDA has also outlined various ways to reduce burdens on sites and participants during the long-term follow-up period; in some cases, it may not be necessary to require much more than an annual phone call to check in with participants.
Bringing a new gene therapy to market is a challenge for many reasons, not the least of which is the litany of clinical trial regulations that can vary from country to country. The European Union has implemented an overarching genetically modified organism (GMO) framework comprised of the Contained Use (2009/41/EC) and Deliberate Release (2001/18/EC) Directives, which together seek to address and mitigate the risks associated with gene therapies and other GMOs. However, individual member states have taken different approaches at implementing these Directives into regulations, which can make already challenging multinational gene therapy trials even more difficult.
The regulatory landscape for gene therapy trials in the United States is established by Department of Health and Human Services (HHS) through the FDA and National Institutes of Health (NIH), with biosafety guidance from other federal entities. The FDA does not distinguish between gene-based and traditional products, requiring an Investigational New Drug Application and Institutional Review Board approval for clinical trials testing new biologics. However, the NIH’s trial oversight framework focuses specifically on trials involving human gene transfer research, which it defines in the NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules (NIH Guidelines) as the deliberate transfer of recombinant or synthetic nucleic acid molecules into a human research participant. The NIH Guidelines also outline various biosafety requirements for research with gene therapies and other genetically modified products that sponsors should consider when planning a new trial.
An important NIH requirement to take into account when planning gene therapy trials that will enroll patients in the United States is that at most sites, the trial must be reviewed and approved by an Institutional Biosafety Committee (IBC) before the study may begin at the site. These committees exist to protect clinical staff, members of the public, and the environment from accidental exposure to the gene therapy product being tested. Accordingly, IBC reviews of gene therapy trials focus on site-level handling procedures, safety equipment (e.g., biosafety cabinets, eyewash stations), emergency response plans, and staff training requirements. Because of this site-specific scope, unsafe work practices described in pharmacy manuals or similar sponsor-provided handling instructions can lead to significant delays in obtaining IBC approval. This is especially true for instructions related to needle handling, decontamination, and waste handling procedures. Wherever possible, sponsors should defer to local IBCs or similar institutional entities for such instructions. Important caveats sponsors should also be aware of are exemptions for certain products, like siRNAs, and the fact that the NIH Guidelines and IBC requirements therein also apply internationally if NIH funds were used to develop the product or run the trial itself. Consultation with biosafety and regulatory experts early on in the study planning process is the best way to ensure gene therapy studies are conducted safely and without unnecessary delays.
Peter Marks, head of FDA’s Center for Biologics Evaluation and Research that oversees gene therapies, earlier this year acknowledged that progress in bringing new gene therapies to patients has been slower than desired and expressed a desire to align global regulations to accelerate the process. While globally aligned regulations will certainly help, nothing will move forward without well-designed, well-controlled clinical trials that demonstrate safety and efficacy. That is no simple task for gene therapy trials, so it will be imperative that sponsors and investigators recognize and avoid the most common problems that have plagued the field thus far.
References
1. European Commission. Rare Diseases, https://research-and-innovation.ec.europa.eu/research-area/health/rare-diseases_en. Accessed 19 August 2023.
2. FDA. Rare Diseases at FDA, https://www.fda.gov/patients/rare-diseases-fda. Accessed 19 August 2023.
3. FDA Long Term Follow-up After Administration of Human Gene Therapy Products | Guidance for Industry. January 2020. https://www.fda.gov/media/113768/download
4. Office of Science Policy, NIH. NIH Guidelines for Research Involving Recombinant or Synthetic Nucleic Acid Molecules. April 2019. https://osp.od.nih.gov/wp-content/uploads/NIH_Guidelines.pdf. Accessed 9 September 2023.

Christopher Doyle, PhD, is Senior Director of IBC Services and an IBC Chair at WCG. He works with clinical trial sites, sponsors, and CROs to ensure research involving gene-based medicines is conducted safely and efficiently. Prior to joining WCG, Christopher worked as a research fellow at the Albert Einstein College of Medicine and Montefiore Medical Center (Bronx, NY), where he explored novel mechanisms of antibody activity against Streptococcus pneumoniae. Christopher received his PhD in Molecular Genetics and Microbiology from Stony Brook University (Stony Brook, NY), where he studied the pathogenesis of Francisella tularensis in a high containment Biosafety Level 3 laboratory.

")





